Portfolio manager summary
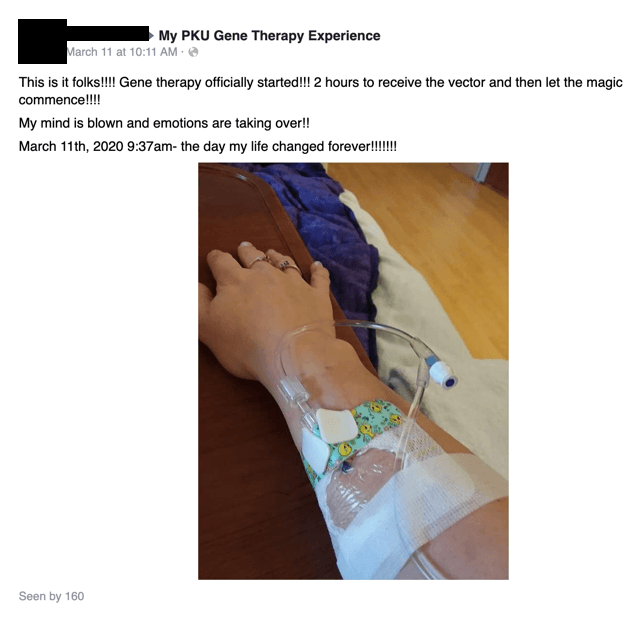
- Y-mAbs (YMAB) is a biotechnology company focused on cancer treatment – it has one FDA-approved drug on the market, Danyelza (naxitamab), which received FDA approval in November 2020 for the treatment of neuroblastoma
- Our diligence reveals that YMAB’s top shareholder is also the same physician in charge of the neuroblastoma program at a major YMAB study site and patient treatment center – his over $100MM ownership in the company is the first time we’ve seen a conflict of interest of this magnitude between patient care/research integrity and financial interests
- We believe YMAB is obfuscating Danyelza’s efficacy – while investor materials have shown objective response rates (ORR) of 68% to 79%, its FDA label shows an ORR of merely 45%; CLVS’s stock plunged 70% in 2015 when it disclosed lower than guided efficacy for an oncology drug
- If this weren’t enough, we believe YMAB’s characterization of highly positive results in frontline trials is disingenuous and a result of selecting the healthiest patients for study – patients with no detectable evidence of cancer
- YMAB received a rare FDA Refusal-to-File (RTF) letter for Omburtamab in October– in the three years ending 2018, just 1% of BLAs received an RTF. We believe this is a sign of execution and cultural issues; management has already missed its own expectations/targets for resubmitting Omburtamab’s BLA
- Investors and/or the SEC might take issue with YMAB’s disclosures about its founder, Thomas Gad; filings disclose the single insolvency of a “personal holding company”, but our review of never before seen Danish corporate filings show at least five entities with no less than 40MM kroner in liabilities that went bankrupt/were dissolved
- While YMAB’s stock rallied from the mid-20s to low-50s from 2019 to early 2021, Gad has sold almost half of his direct holdings
- We believe that the combination of Danyelza’s “real” efficacy and YMAB’s poor disclosure presents significant risk to investors, and that Danyelza will not be as successful as the company and sellside would like you to believe – we assign a $16 price target to the stock, down ~55% from current levels
Overview
YMAB is a biotechnology company focused on the development of antibody-based products for the treatment of cancer. YMAB’s product portfolio was licensed from Memorial Sloan Kettering Cancer Center (MSKCC), and its only FDA approved product, Danyelza (naxitamab), is for the treatment of relapsed or refractory (R/R) high-risk pediatric neuroblastoma. Neuroblastoma is a rare, almost exclusively pediatric cancer that develops in the nervous system. Unfortunately, this disease is “associated with poor long-term survival” and accounts for 15% of pediatric cancer deaths.
Obviously, treating pediatric cancers is an exceptionally noble mission, and we applaud YMAB and the team at MSKCC for their work. What is not noble, however, is what appears to be a pattern of unusual and incomplete disclosure about drug efficacy and management history, execution issues, and stock sales.
As an example of the above, we cannot reconcile why YMAB would tell investors that Danyelza’s efficacy exceeds 75%, when its recent FDA approval shows an efficacy of just 45%. Read on to learn why we assign a $16 price target to YMAB, down ~55% from current levels.
Part I: A disclosed and disturbing conflict of interest
Before going into the substance of our thesis, we’d like to bring up an issue which we believe is a significant public health and governance issue.
A look at YMAB’s holders list shows that its top holder is Nai-Kong Cheung:
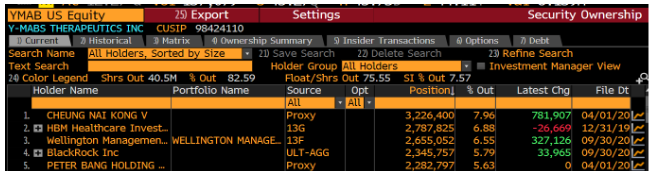
Who is Nai-Kong Cheung? Dr. Nai-Kong Cheung, who owns over ~$100MM in YMAB stock, is the head of the neuroblastoma program at Memorial Sloan Kettering (MSK) which is not only a study site for YMAB’s drugs, but also a treatment center for neuroblastoma patients.
Said another way – Dr. Cheung, through his massive stock ownership (likely well in excess of his MSK compensation), has a clear financial interest in trial outcomes and patient care at MSK. YMAB licensed its drug portfolio from MSK and granted shares to Cheung for “his involvement in the development of technology licensed from MSK in consideration for his services with respect to such technology development.”
We believe that this, despite any policies and procedures, creates a massive conflict of interest that could favor YMAB over patient care (choosing a YMAB product for reasons outside of clinical superiority, for example) or in clinical trials (like presenting data in a favorable manner). WE ARE NOT ACCUSING DR. CHEUNG OF ANYTHING UNTOWARD, BUT THE OPTICS HERE ARE TROUBLING.
Bear the latter in mind as we discuss certain data reporting issues in the next section.
This would not be the first time conflicts of interest have touched MSK – in 2018, ProPublica and the New York Times found that “…several top executives and board members [at MSK] had profited from relationships with drug companies, outside research ventures or corporate board memberships.

Source: NYT
These findings led MSK to conduct a review, which found:

Source: ProPublica
We believe that MSK’s previous cultural issues, Cheung’s significant ownership in YMAB, and his leadership position in the very department conducting patient care and research using YMAB drugs creates an alarming set of conditions.
The findings we share below, particularly around the efficacy discrepancies between how YMAB has presented its drug data compared to its FDA label, should be considered in light of the conflict of interest mentioned above.
Part II: Danyelza’s different data for different audiences
After the close on 11/25/20, YMAB announced that YMAB’s Danyelza (naxitamab) received its FDA approval for relapsed/refractory pediatric neuroblastoma. The resulting FDA label highlighted what we believe to be significant inconsistencies in the efficacy data that YMAB has presented to both the market and the medical community.
Before we move on to laying out this data, let’s first recap some important terminology related to the efficacy of oncology treatments:
- Objective response rate (ORR): according to the FDA, ORR is “the proportion of patients with tumor size reduction of a predefined amount and for a minimum time period”
- Complete response (CR): according to the FDA, CR is defined as “as no detectable evidence of tumor”
- First line, or frontline treatment – the initial, or primary treatment recommended for a disease
- Relapsed/refractory disease (R/R) – setting where the disease does not respond to or gets worse on therapy or after showing a response to prior therapy
These terms, along with progression-free survival (PFS), which is defined as the “time from randomization until objective tumor progression or death”, are critical in allowing physicians and other important stakeholders to compare treatment options.
Now, let’s walk through what we believe are significant inconsistencies in how YMAB presented Danyelza’s data.
Shot: Data per Company Investor Deck
In YMAB’s December investor deck, YMAB shows that Danyelza, in Study 201, which was the dataset submitted to the FDA for its biologics license application (BLA), has an ORR of 79% and a CR of 71%:
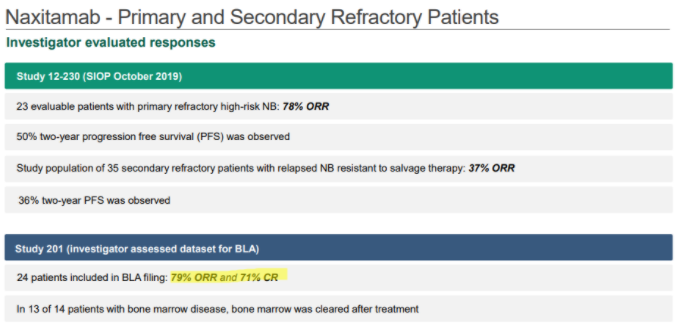
Source: YMAB December Investor Deck
Shot: Data per SIOP Poster
In a poster presented at the International Society of Paediatric Oncology’s October 2020 Virtual Congress (SIOP), YMAB shows a lower ORR of 68% and a CR of 59%:
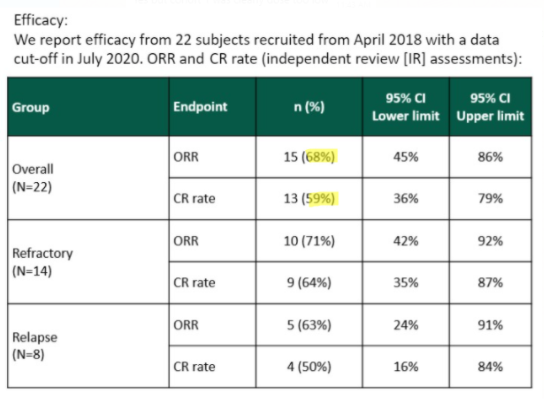
Source: SIOP 2020 poster screenshot
It is notable that YMAB has changed to presenting the SIOP data in its January 2021 and subsequent investor decks, without explanation:

Chaser: Data per FDA Label
And then there are the efficacy metrics found in the FDA label (approved November 2020) for Danyelza, which we believe is the most credible source and the one most likely to be relied on by physicians, showing a dramatically lower ORR of 45% and CR of 36%.
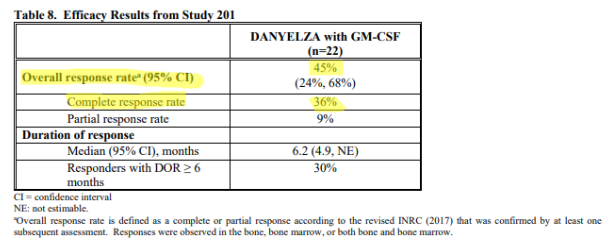
Source: FDA label
To put this in context, the FDA label ORR and CR are 34 percentage points and 35 percentage points, respectively, LOWER than what YMAB presented in its investor deck – and 23 percentage points for both metrics LOWER than what was presented at SIOP:
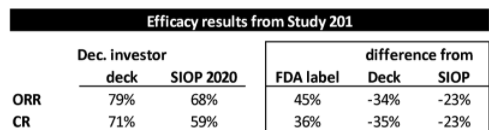
Source: Investor deck, SIOP 2020, FDA label
What gives?
Why would YMAB show investors and the medical community efficacy metrics dramatically higher than those that ultimately blessed by the FDA? Did YMAB analyze the data differently than the FDA would? If so, why?
It is our view that YMAB chose to interpret and present the data in a manner that was favorable to them, and, by extension, to the stock price.
Physician commentary from a pediatric oncologist at a large neuroblastoma treatment center suggests the investor and SIOP data was limited to a single site:
“Single site versus multi-site. That [higher number] is that two decades of data that they have for patients who were treated at Sloan Kettering as a single institution. This goes back to how much cherry-picking occurred.”
YMAB’s own explanation is unsatisfactory:
“Well, we have 3 sets of data out from that study. We have the investigators’ response evaluation, which shows about 79% overall response. We have the independent tumor response evaluation, where we have 2 independent radiologists and 2 independent pathologists looking at bone marrows and scans from the patients and concluding that 68% of the patients is responding. And the same data set was sent to the FDA, and their decision was that they excluded a number of our responders for various kinds of reasons. And I could have spent the next 3 months discussing whether it was fair and reasonable and added additional information on data. I felt it was more important to get the treatment out to the patient than having that discussion that the FDA wanted to remove a number of our responders out of the first 24 patients.”
Source: Call on 12/16/20
The FDA is clearly using a different evaluation criteria than YMAB, and investors are expected to just believe that YMAB’s interpretations are better than the ultimate arbiter of drug safety and efficacy? This, in our view, is not a good look.
It’s like Clovis Oncology, in a bad way
More tangibly, YMAB is bringing to market a product whose label efficacy is dramatically lower than what YMAB initially communicated. This is something investors should be concerned about – Clovis Oncology found itself in a similar situation, to the detriment of its shareholders. After telling investors its rociletinib showed a 59% response rate in lung cancer, it provided a November 2015 update revising the number down 25 points to 34% response rate. This sent Clovis’s stock down ~70% in one trading day.
Danyelza’s FDA label efficacy is more than 30 points below what YMAB was telling investors.
We have doubts about efficacy and success
Along with the company credibility issues that this implies, it means to us that Danyelza will struggle to compete against incumbent United Therapeutics’ Unituxin (dinutuximab) in the R/R setting. In this paper, published in The Lancet, we learn that Unituxin has a median PFS (defined as how long a person lives without their disease worsening) of 2.1 years for R/R neuroblastoma:
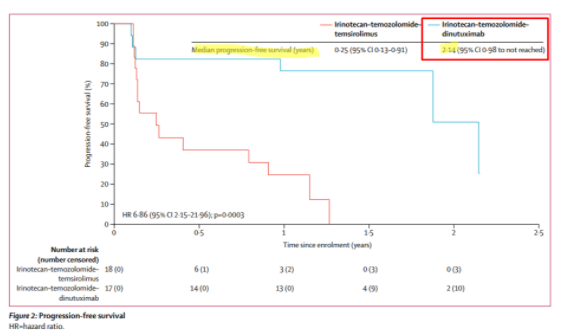
Source: The Lancet
For Danyelza, YMAB uses an analogous metric known as Duration of Response (DoR), which is effectively PFS for patients that have responded to the treatment, a more favorable metric since it excludes patients that failed to respond. Danyelza’s DoR is just 6.2 months:
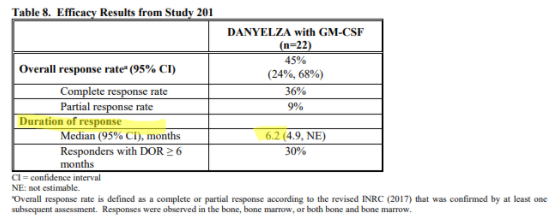
Source: Danyelza FDA label
To put this in perspective, competitor Unituxin has a progression free survival, which includes non-responsive patients, of 2.1 years, while Danyelza’s DoR (which EXCLUDES non-responsive patients) is just 6.2 months.
The implication is that Unituxin’s DoR should be greater than 2.1 years, and that Unituxin has an over 4x better PFS or DoR than Danyelza.
Physicians will ultimately decide how they want to use the drug, but we are bearish on its prospects outside of MSK.
The same oncologist we quoted earlier said that “no one” outside of MSK has been able to make their own evaluations of Danyelza’s study data. Further, due to the data issues we highlighted, comparability of Danyelza to other drugs was characterized as “nebulous”:
“No one had an opportunity to to make their own evaluations if you weren’t at Sloan Kettering. Even if you referred a patient there, it was still a little bit nebulous as to how does this data compare.”
If you test healthy patients, you get good results
YMAB has also been promoting Danyelza’s results in the frontline setting, where it would compete with Unituxin’s on-label use. In YMAB’s materials, they show a 2-year event-free survival rate of 74.3%, comparing favorably to Unituxin’s 63%:
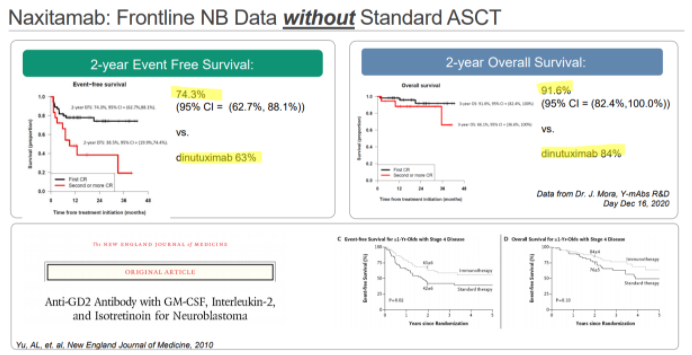
Source: Investor deck
For anyone that reads this, it’s absolutely amazing to see this kind of EFS without the use of the standard of care stem cell transplant. And we think that’s exactly the reaction we think YMAB wants you to have.
What they don’t tell you is buried in disclosures presented during SIOP – the patients in this study were in complete remission (i.e., they had no detectable evidence of cancer):

Source: SIOP
The boxed areas say “…improved outcomes of HR-NB patients in complete remission (CR) after induction therapy and autologous bone marrow transplant (ABMT)” and “Eligibility criteria included non-evidence of disease”.
So Danyelza was given to patients with no evidence of cancer after receiving chemo, and the results of that study were good? That should come as no surprise.
In the case of Unituxin, the eligible patients in the study were not limited to those only in complete remission:

Source: NEJM
To put this simply – YMAB is comparing the results of a drug given to those with no detectable evidence of cancer to the results of a drug given to sicker people. This, in our view, is disingenuous.
We believe this is a problem in study design – by using the healthiest of cancer patients, how could the results NOT come out positively? How can these results be taken credibly?
Why on earth would a physician choose to prescribe Danyelza? Not only did YMAB’s efficacy come in lower than it presented, but the survival benefits appear to be dramatically lower in R/R when compared to Unituxin. Not to mention the patient selection for Danyelza in frontline seems highly biased.
In our next section, we highlight YMAB’s execution issues for two of its drugs, Omburtamab and Nivatrotamab.
Part III: Execution issues
Omburtumab’s refusal to file as a cause for concern
YMAB first landed on our radar in October and the reason that it attracted our attention was something that rarely happens in the biotech world. The company received an FDA Refusal-to-File (RTF) letter for the BLA for its pipeline drug Omburtamab.
An RTF letter is a notice from the FDA that a submitted drug application is incomplete, and that the FDA is refusing to accept the application until the sponsor corrects the identified issues.
RTFs are relatively rare – between 2010 and 2017, the FDA issued only 73 RTF letters – over that same period, 1,042 new drug applications and BLAs were filed, implying that just 7% of applications were deemed sufficiently incomplete as to warrant an RTF letter. More specifically, in the three years ending 2018, less than 1% of submitted BLAs (most relevant to Omburtumab) received an RTF.
The FDA takes the view that issues that are addressed by RTFs are known as “complex significant deficiencies”, note that these are the FDA’s words, not ours and they include:
- Applications that are materially lacking to the extent they would not permit efficient review
- Parts of an application that contain inadequate information
- Reliance on a single trial when prior communication with FDA determined the need for more than one trial
- Failure to submit assessment of studies related to the potential abuse of a drug
- Content not submitted electronically where the FDA has specifically requested electronic format
In our view, these seem like basic blocking and tackling for any drug company, and the inability to comply with them is a cause for concern. In the case of YMAB, detail around the RTF is scant, but sufficient enough, in our view, to call into question the company’s ability to execute. In its RTF release, YMAB said that the “FDA determined that certain parts of the Chemistry, Manufacturing and Control (“CMC”) module and the Clinical module of the BLA require further detail”.
A subsequent conference call with investors was similarly vague, seeming to chalk up the CMC module issue to YMAB “anticipating that the FDA would have been okay” with what the company provided:
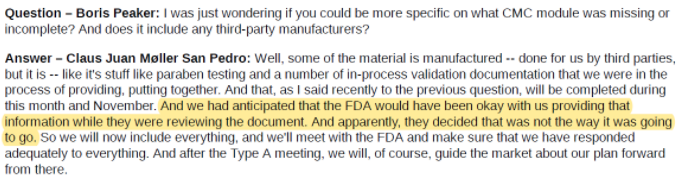
Source: Investor update call on 10/6/2020
On the clinical side, it appears that FDA is asking YMAB for tumor response data independently evaluated according to the RANO criteria, an industry standard set of criteria published in 2010 used to assess response to first line treatment:

Source: Investor update call on 10/6/2020
To us, this reads like YMAB simply chose not to provide independent data evaluated according to a widely accepted framework.
The more galling part is that FDA “found out” that YMAB had the data needed to do this:

Source: Investor update call on 10/6/2020
Reading further, we learn from YMAB’s CEO that the study data was, in fact, not validated and that the FDA had allowed them to provide the RANO criteria evaluation as a post-marketing commitment:

Source: Investor update call on 10/6/2020
This, in our view, strains credulity. The FDA was going to accept an application where the data are not independently evaluated and then changed its mind?
As if that weren’t enough, CEO Moeller mentions on this call that they would work to resubmit Omburtamab’s BLA “before the end of 2020”:

Source: Investor update call on 10/6/2020
But in 4Q20 results, that timeline got pushed to the end of the second or third quarter of 2021, suggesting the problems hinted at during the October 6 investor call are perhaps thornier than disclosed:
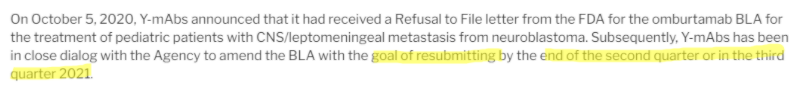
Source: Press release
We think the fact that YMAB did not provide data evaluated under RANO criteria as an indicator of company credibility and execution. This is, in our view, entirely consistently with the Danyelza efficacy story we shared above.
In its 2018 Report on the State of Pharmaceutical Quality, in noting how it has processes to refuse poor-quality drug application submissions, FDA discusses drug product application quality, noting that “[a]nother important indicator of the overall State of Quality is the quality of drug product applications sent to the FDA, which may reflect a firm’s quality culture across its operations.” (our emphasis)
The hat trick – issues with a third product
On December 16, 2020, YMAB released a pipeline update that included commentary on the aforementioned Danyelza and Omburtamab, as well as a third drug candidate, Nivatrotamab.
In the case of Nivatrotamab, we learn that no complete or partial responses were achieved in YMAB’s 10 patient study, but they are still preparing for a Phase 2 study:

Source: Press release
So in addition to Danyelza’s data issues and Omburtamab’s RTF, we have Nivatrotamab’s poor initial results. This collection of events suggests significant execution and cultural issues, which, when combined with YMAB’s incomplete management disclosure and founder’s stock sales, makes us very negative on the company.
Part IV: YMAB’s disclosure issues appear to extend to its founder
Thomas Gad is YMAB’s “Founder, Chairman, President, and Head of Business Development and Strategy and Director.”
In the proxy, there is a curious mention of a Y-mAbs Therapeutics ApS (“Old YMABS”), a Danish company explained as Gad’s “personal holding company involved in research and development activities in the pharmaceutical industry.” The proxy goes on to tell us that “the company was placed in liquidation proceedings in 2015. The company is now under compulsory dissolution proceedings.”
Taking this at face value, it seems innocuous. But, as we have seen, YMAB appears to have an issue with full and complete disclosure, and this, unsurprisingly, extends to the company’s disclosures about its founder.
Our examination of Danish corporate records found not just Old YMABS’s bankruptcy, but four other Gad-affiliated companies that were dissolved/went bankrupt, and a sixth company which recently absorbed Old YMABS.
Here is an overview of Gad’s corporate activity:
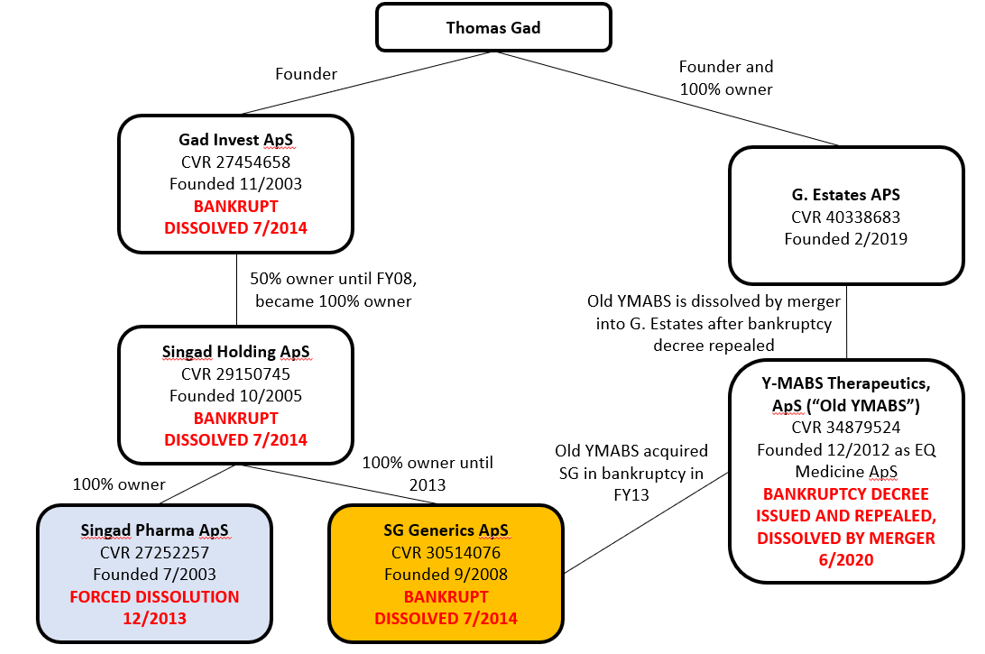
Sources: Danish Corporate Filings for Thomas Gad, Gad Invest, Singad Holding, Singad Pharma, SG Generics, Old YMABS, G. Estates
What is most impressive to us is that it appears that there are two separate insolvency storylines here.
- The first is the insolvency of Singad Pharma, whose activities consisted of importing and distributing pharmaceuticals (translated from the Danish corporate filings). As you can see below, of Singad Pharma, Singad Holding, and Gad Invest, Singad Pharma was the only entity with real revenue and profit. But it wasn’t very profitable – for the period which we were able to obtain filings, its best operating margin was in 2008 at 3.75%, when sales peaked. Between 2008 and 2011, Pharma’s sales fell 65% but liabilities grew 165%, and the equity value went deeply negative to -27MM kroner. We believe that the insolvency of Gad Invest and Singad Holding were the result of Singad Pharma’s clear insolvency:
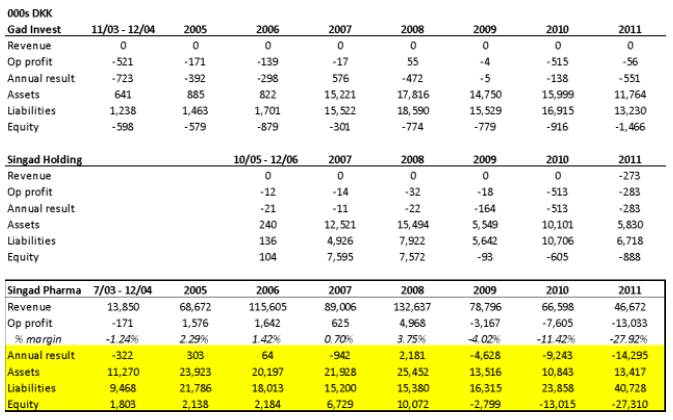
Sources: Danish Corporate Filings for Gad Invest, Singad Holding, Singad Pharma
- SG Generics appears to be another story. Founded sometime in FY2008, around the same time when Gad Invest became a 100% owner of Singad Holding, SG Generic’s business was also the import and distribution of pharmaceutical products (translated from Danish corporate filings). While we only have three annual filings for SG, it’s quite clear that the business never went anywhere – producing no revenue in the 2008 to 2011 period:
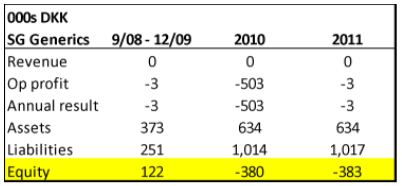
Source: Danish Corporate Filings for SG Generics
In August of 2013, a request for dissolution was sent to the probate court in Elsinore, and in May 2014 a bankruptcy decree was handed down:

Source: Danish Corporate Filings for SG Generics
Curiously, in December 2012, Gad formed another entity, EQ Medicine ApS, which was renamed Y-MABS Therapeutics ApS (what we called Old YMABS). In Old YMABS’s 2013 filing, we learn that it acquired SG Generics in bankruptcy, and that SG’s condition wouldn’t affect Old YMABS’s business:

Source: Old YMABS’s 2013 filing and Google Translate
But clearly it did affect Old YMABS, because in 2016, a request for dissolution was filed, and a bankruptcy decree was handed down. The decree was subsequently repealed and Old YMABS was dissolved by merger in June 2020 in yet another Gad entity, G. Estates, formed in 2019:


Source: Old YMABS’s 2013 filing
So in the SG Generics story, we have Gad forming two entities (Old YMABS and G. Estates), which subsequently absorbed two bankrupt entities (SG Generics and Old YMABS).
The most galling fact here is that YMAB only discloses to its investors the bankruptcy of Old YMABS, which had just 81k kroner of assets and 9k kroner of liabilities at the end of 2014. Investors are left completely in the dark about the chain of insolvency beginning with Singad Pharma, which had an 41MM kroner in liabilities. These are undisclosed insolvencies and dissolutions never before seen by investors.
Why was this left out?
It is our view that this is yet another significant data point in a pattern of poor and shifting disclosure designed to sweep unfavorable events and data under the rug.
Part V: Founder’s stock sales
Since April of 2019, Thomas Gad has amassed a breathtaking amount of stock sales, regularly selling every couple weeks as the stock went from the $20s to the low $50s. In April 2019, we estimate that Gad directly held 1.19MM YMAB shares – as of March 3, 2021, Gad holds 596k shares, implying that he sold ~50% of his ownership since 2019. Inclusive of sales following option exercises, this amounts to 795k shares!
All the while, investors bid the stock from the mid $20s to the high $40s.
It seems rather concerning that while the sellside salivates over the company and management continue to promote YMAB that Gad himself is dramatically reducing his exposure to the company he founded:
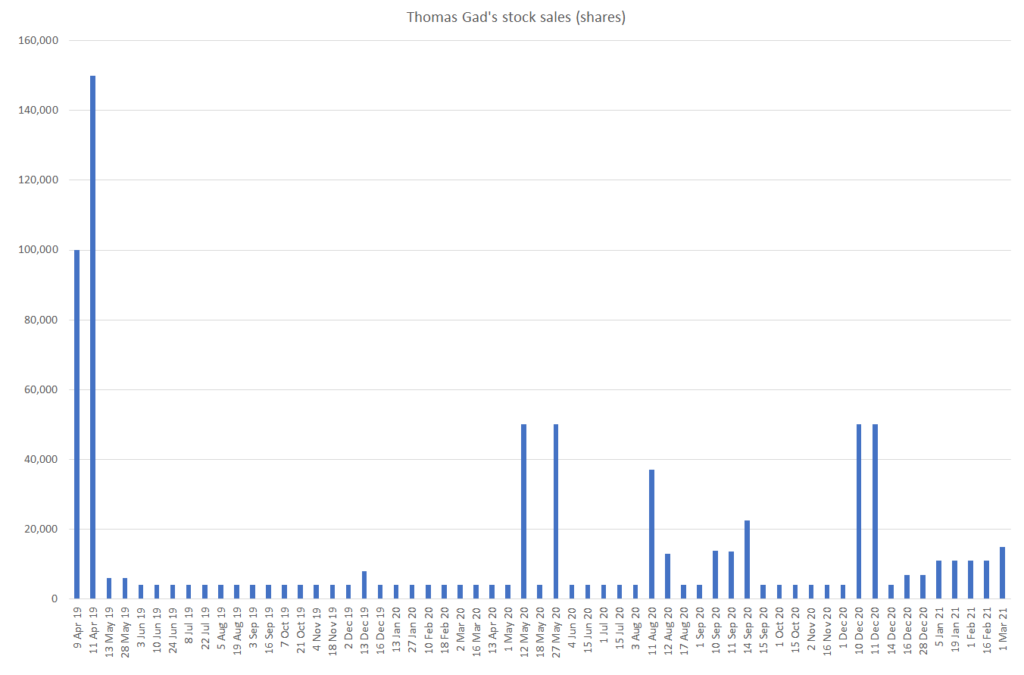
Source: SEC
We estimate that this has resulted in net profits to Gad of approximately $27.3MM: $8.3MM in 2019, $17MM in 2020, and $2MM in 2021 year to date.
Why should investors continue to hold the stock if the founder and Chairman is lightening up his ownership?
Part VI: Conclusion and valuation
The Mariner Instant Replay shows that:
- From a public health perspective, Dr. Cheung’s ownership of over $100MM YMAB stock is a conflict of interest that could be to the detriment of both patients and investors
- YMAB’s Danyelza appears to have an efficacy and disclosure issue – while showing investors and physicians efficacy ranging from 68% to 79%, Danyelza’s actual label shows efficacy of just 45%
- Further, YMAB appears to have tested Danyelza on patients with no observable cancer, making the high efficacy numbers in the front line setting almost meaningless
- YMAB’s Omburtamab RTF is suggestive of poor execution and data issues
- YMAB’s Nivatrotamab doesn’t appear to show any efficacy thus far
- YMAB has failed to disclose that Thomas Gad’s bankruptcy history is not just limited to his “personal holding company”
- Gad has unloaded stock at a breathtaking pace, having sold ~50% of his ownership since 2019
We believe that YMAB is obfuscating its Danyelza efficacy and its Omburtamab execution. The sellside expects Danyelza to take almost 40% of the R/R neuroblastoma market in the long run. We just don’t see it.
It is our view that Danyelza will never reach more than 20% patient penetration, and that, despite sellside estimates to the contrary, Omburtamab will not see revenue until 2024.
Under these assumptions and 12.5% discount rate in a DCF, we arrive at a price target of $16, down ~55% from the most recent close.